编译:微科盟小狗尾巴,编辑:微科盟木木夕、江舜尧。
导读
背景:家畜肠道微生物群组成的分析可能有助于调节微生物复杂表型。然而,有关猪肠道微生物群落的宿主遗传控制我们知之甚少。以往的研究是基于经典的“单基因-单性状”方法分别评估宿主基因组控制肠道原核生物和真核生物群落的作用,忽略了细菌和原生生物群落之间的串扰及基因间相互作用对猪肠道微生物生态系统的影响。为了解决这一空白,我们提出了一种关联权重矩阵方法来生成一个基因共关联网络,将重点放在负责宿主细菌和原生生物群落之间串扰的基因调节子上。
结果:为了确定宿主基因控制健康猪微生物群落多样性和组成的能力,我们对来自390头猪的39个微生物表型进行了全基因组关联研究(GWAS),其中包括2个多样性指数,以及31个细菌属和6个共生原生动物属的相对丰度,对390头猪进行了70K 单核苷酸多态性(SNP)基因分型。GWAS的结果通过一个三步分析管道进行处理,包括(1)关联权重矩阵;(2)调控影响因子;(3)偏相关和信息理论。推测的基因调控网络由3561个基因(与相关的SNP-P<0.05)与738,913个连接(SNP之间的共同关联)组成。本研究结果表明了猪肠道微生物生态系统的复杂性和多基因特性。在该网络中有5个突出的调节子,分别是PRDM15、STAT1、SSC-mir-371、SOX9和RUNX2,且分别聚集了942、607、588、284和273个连接。PRDM15可调节WNT和MAPK-ERK信号上游调节因子的转录,以保护天然的多能性,还可调节Th1和Th2型免疫应答的产生。信号转导分子STAT1长期以来一直与免疫过程相关,最近被确定为猪繁殖与呼吸综合征疫苗反应的潜在调节因子。本研究丰富了免疫相关途径的调控因子数量,预测靶点包括先前报道的与猪、小鼠和人的微生物区系图谱相关的候选基因,如SLIT3、SLC39A8、NOS1、IL1R2、DAB1、TOX3、SPP1、THSD7B、elF2、PIANP、A2ML1和IFNAR1。此外,我们还证明了宿主的遗传变异与产生丁酸盐的细菌相对丰度和宿主性能有关。
结论:综上所述,本研究结果确定了与宿主微生物群调控相关的调节因子、候选基因和机制,并进一步强调了分析方法的有效性,通过利用细菌和原生生物之间的多效性和串扰在宿主-微生物组相互作用中发挥重要作用,确定了可以改善宿主性能和微生物特性并且可以纳入育种计划的遗传标记和候选基因。
原名:A gene co-association network regulating gut microbial communities in a Duroc pig population
译名:调控杜洛克猪肠道微生物群落的基因共关联网络
期刊:Microbiome
IF:14.650
发表时间:2021.02.21
通讯作者:Yuliaxis Ramayo-Caldas
通讯作者单位:西班牙巴塞罗那农业食品研究与技术研究所

表1列出了受GWAS影响的39种表型在三个名义P值阈值(<0.001、0.01和0.001)下的显著SNP数量(SNP)和错误发现率(FDR)。结果表明在P值越小时,SNP和FDR之间存在取舍关系,即:在P <0.05、0.01和0.001,平均显著SNP(平均FDR)数值分别为4015.3(FDR =50.6%)、272.8 (FDR =16.5%)和87.6 (FDR=5.8%)。结果显示P <0.001时,Faecalibacterium的SNP最高(N=381;FDR=1.1%);相反,Catenibacterium的SNP最低(N=25;FDR=17.0%)。表2列出39个表型中最显著的SNP的基因组图谱位置和统计关联强度,并表明每对SNP-表型的最近基因距离和同一性。在5个表型中,8号染色体包含关联的SNP最多,包括位于SorCS2的编码区(SNP Rs320095924)和位于TRIM2的编码区(Rs329143797)。
表1. 三个P值下39个表型的显著SNP数(N)和错误发现率(FDR,%)。

表2. 39个表型中最显著SNP的基因组图谱位置和关联强度。
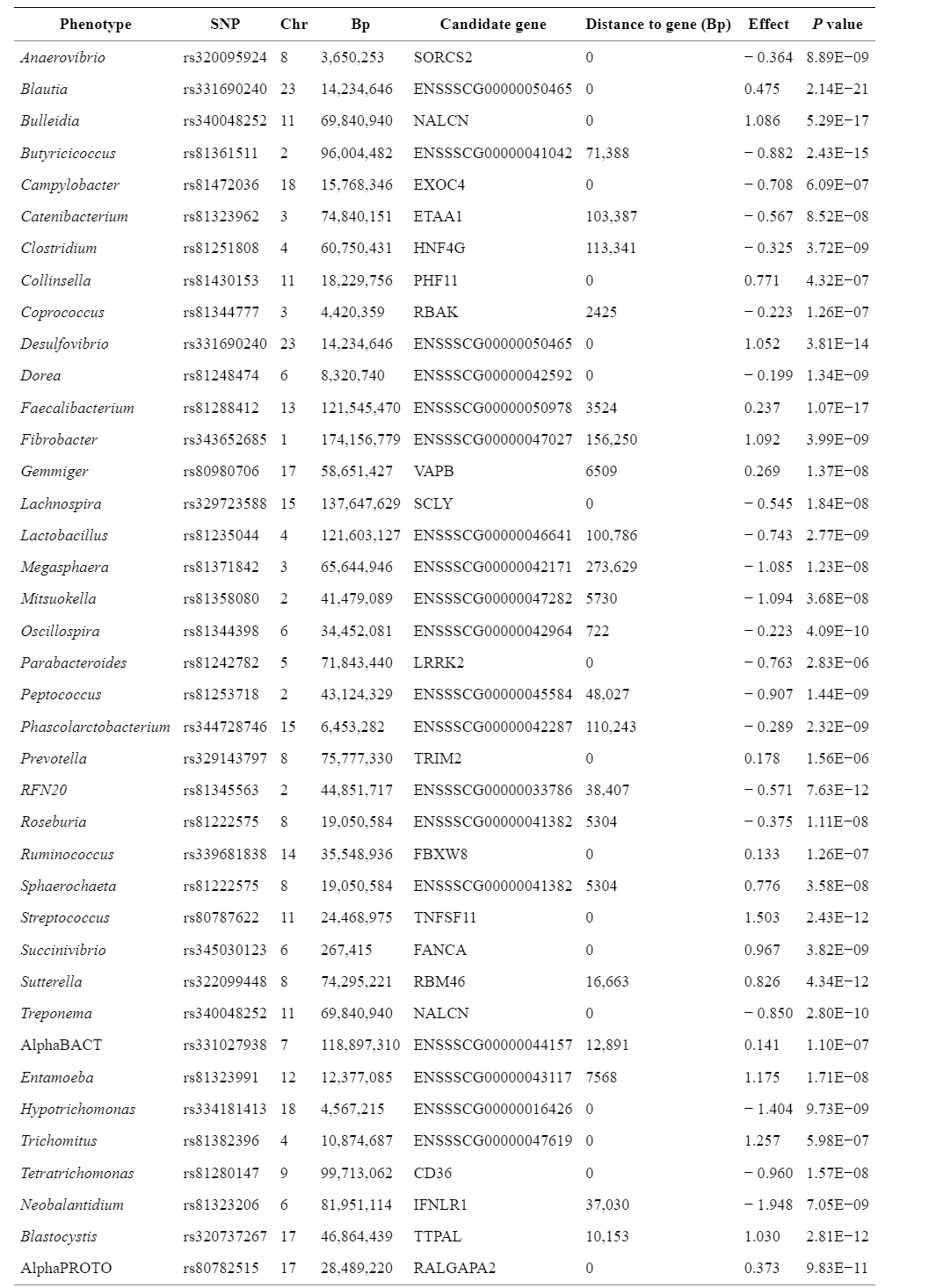
GWAS的结果显示基于关联权重矩阵法(AWM)、基因共关联网络和调控影响因子方法的基础。在第一步中,通过对39种表型SNP效应的预估,基于本研究中“方法”一节里描述的分析管道,得到了在单个基因上的3,561个SNP组成的AWM,其中121个被注释为TF基因和7个microRNA基因。此外,调控影响因子(RIF)分析显示47个关键调控因子(包括3个microRNA基因),但其中10个与任何表型都没有显著关联(P < 0.05),其相关性被GWAS忽略。在剩余的3,551个基因中,相关表型的数量为1至13,平均为3.79。有84.08%的SNP位于基因内,15.92%位于注释基因的上游/下游。使用AWM计算微生物性状之间的相关性(跨细菌、原生生物的多样性和丰度的标准化SNP效应),并将其可视化为分层树簇,其中强正相关和负相关分别显示为接近度和距离(图1)。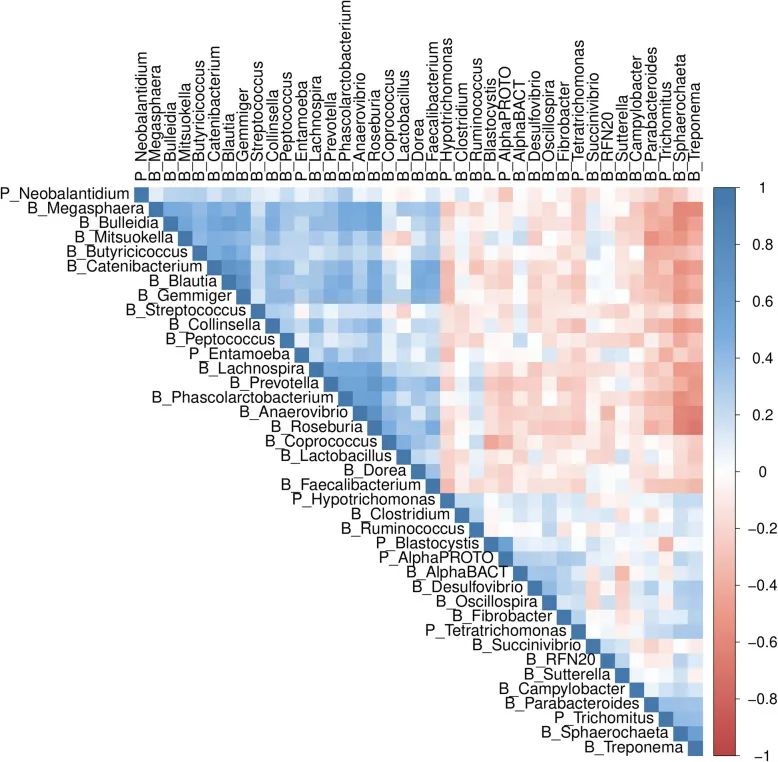
图1. SNP共关联关系:基于AWM中包含的3,561个SNP基因的32个细菌(前缀为“B_”)和7个原生生物(前缀为“P_”)表型的关联矩阵热图。补充图1显示了在AWM分析中包含的3,561个SNP-基因的偏相关和信息论(PCIT)推断的基因共关联网络的概况,该网络由738,913条边连接,其中374,116条为阳性,364,797条为阴性。网络图中带颜色的节点表示具有关联最强的表型(补充图1)。该网络的特点是中央模块包含了大多数细菌与原生生物存在的联系,四周较小的模块大多是表型特异性的。AWM分析显示仅有少数基因是细菌和原生生物α多样性的关键表型:细菌241(深蓝色节点)和原生生物231(红色节点)。相比之下,其他细菌共包含2,568个基因(浅蓝色节点),而其他原生生物丰度表型共含有521个基因(橙色节点)。表3和图2显示了RIF分析从AWM分析选择的基因中重新鉴定了47个关键调节因子,较突出的是PRDM15、STAT1、ssc-mir-371、SOX9和RUNX2,分别聚集942、607、588、284和273个连接(表3)。PRDM15是最有价值的调节因子,与10个特征有关(表1)。PRDM15是一种锌指序列特异性染色质因子,调节WNT和MAPK-ERK信号传导的上游调节器的转录,以保护原始态多能性,并调节TH1和TH2型免疫反应的产生。类似地,据报道,MicroRNA ssc-mir-371在猪中的多能性调控中发挥着重要作用。另一方面,已发现肠道无微生物的小鼠分别通过SOX9和RUNX2的中介产生肝损伤和骨质损失。信号换能器STAT1与五个微生物特征有关(表3)。STAT1长期以来一直与免疫过程有关,最近被确定为猪繁殖与呼吸综合征疫苗反应的潜在调节因子。值得注意的是,预测靶基因的32和22.5%之间分别具有至少一种TF结合位点,分别为STAT1和PRDM15(补充表2)。MicroRNA ssc-mir-371被鉴定得到155个结合位点(补充表3),其中有71个不同的mRNA基因(初始588个共同相关基因的12%)。在反向互补的3′-UTR中搜索随机miRNA结合位点,总共得到115个不同的结合位点(补充表3),包括48个不同的mRNA基因,即与背景随机miRNA mRNA相互作用相比,ssc-miR-371种子中miRNA 7mer-m8结合位点的预期数量增加了1.48倍。当我们评估rs320008166(n.59T> C)存在的假定结构后果时,前体miRNA发夹折叠的最小自由能(MFE)降低。具体来讲,在miRNA前体区域第59位的这个备选C等位基因的存在意味着野生型miRNA序列中G:U摇摆配对更稳定,引入了稳定的典型Watson-CrickG:C配对。而携带T等位基因的miRNA发夹的MFE = -35.44千卡/mol, T等位基因的存在意味着估计的MFE = -37.74千卡/mol。总体而言,与前人最初发表的AWM研究一致,本研究提供了一些预测的TF靶基因的电子验证的启动子序列,也是首次提供预测的miRNA靶基因的启动子序列。表3. 调控影响因子(RIF)分析揭示的47个关键调节因子的清单及其RIF1和RIF2得分,与细菌(Alpha_B)和原生生物(Alpha_P)多样性的标准化关联,最强关联的表型,多效性,以及PCIT推断的网络中的连接数量。

图2. 基因共结合子网络:PCIT推断的基因共结合子网络,由RIF(方块)确定的47个关键调控因子和它们的第一个邻居(椭圆)组成,具有很强的显著相关性(> 0.7)。节点颜色被映射到关联性最强的表型:深蓝色代表细菌α多样性(Bact_Alpha),浅蓝色代表其他细菌丰度表型(Bact),深绿色代表原生质体α多样性(Prot_Alpha),浅绿色代表其他原生生物丰度表型(Prot)。粉色和青色边分别表示正相关和负相关,节点大小表示多效性的大小。表3中的值可以发现一些有趣的关系。多效性和连通性显著相关(r = 0.571;P < 0.0001),表明这两个指标都是调节因子多潜能能力的指标。这一发现具有相关性,因为虽然多效性是从GWAS中显著表型的数量计算出来的,但连接的数量是共同关联网络中连通性的一个特征。两个截然不同的概念,当连通性和多效性指向相同的结果时,就突显了调节因子的相关性。同样,虽然RIF1和RIF2中度相关(r = 0.421,P < 0.001),但只有RIF2与多效性显著相关(r = 0.471,P < 0.001),而且连接性更显著(r = 0.806,P < 0.0001)。据我们所知,这种以前从未记录过的关系表明,可根据RIF2的分数优先考虑调节因子的能力,在我们的案例研究中,通过区分与细菌特性相关的基因和与原生生物表型相关的基因(即在开发RIF时使用的对比),RIF2分数具有一种巧妙的能力来解决“细菌与原生生物”的串扰。虽然α多样性指数是捕获AWM基因的关键表型,但为了探索“细菌与原生生物之间的串扰”,我们进一步识别了具有多效性潜力的基因。图3显示了基因与细菌中的α多样性、原生动物的α多样性及其在AWM中包括的3,561个SNP基因之间的多效性之间的三向关系。该图表明,实际上与α多样性表型关联接近零的SNP基因在反映其潜在多效性的其他大量微生物丰度表型中仍显著相关。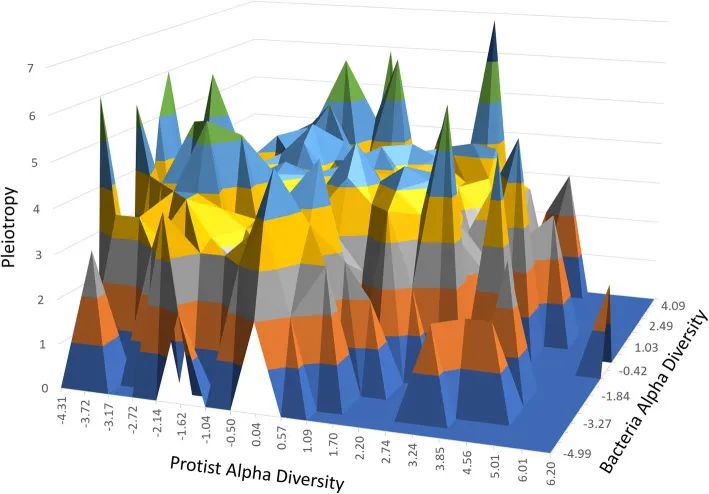
图3. 微生物群落多样性和多效性。原生生物(宽度)、细菌(深度)及其多效性(高度)的SNP与α多样性之间的3向关系的曲面图,测量的是显著相关的原生生物和细菌属的数量(调整后的P值<0.05)。在这项研究中,我们提出了一种系统生物学的方法来识别与39个微生物性状相关的候选基因、调节因子和生物途径。基于预估加法值的集群分布反映了不同的群落组成类型(肠型集群)以及猪肠道微生物区系的已知共生模式。例如,猪肠道肠型驱动分类单元Prevoella和Mitsuokella聚在一起,并远离包括密螺旋体和乳球菌的第二个集群(图1)。我们还注意到可以产生丁酸盐的菌属,如Faecalibacterium,Dorea,Blautia,Butyrococcus和Coprococcus倾向于紧密聚集,这表明加性值具有共同的方向性,并且可能是这组分类群的共同遗传控制。用短读测序平台靶向16S rRNA可变区不能实现辨别物种水平的分类,结果表明本研究提出的分析方法对恢复肠道微生物生态系统的关键生态特性是有用的。基因共关联网络(补充图1)揭示了预测目标基因的同一性,以及猪肠道微生物生态系统多样性和组成的更高复杂性和多基因性质。值得注意的是,已有研究证实了猪宿主基因组与Crespo等人2019年报告的六个细菌属的相对丰度之间的关联(补充表4)。此外,本研究还发现最近报道的68个基因中有27个基因与猪肠道微生物组成员的α多样性和相对丰度相关(补充表5)。值得注意的是,Bergamaschi等人2020年的研究中通常鉴定的基因包括PRDM15。如前所述,尽管遗传背景、年龄、饮食和其他环境因素的研究之间存在差异,但PRDM15与最高数量的性状(表3)和显示最高营养价值的调节剂相关。已确认的关联包括Crespo等人2019年报告的17个QTL中的11个(补充表4),以及Bergamaschi等人报告的与Clostridium, Succinivibrio, Bacteroides, Prevotella,Blautia, Turicibacter, Treponema, Mobiluncus和 Oscillibacter成员相关的QTL。因此,我们的结果表明,与人类和小鼠中进行的宿主基因组-微生物群关联相反,在猪的研究中报道的几个QTL可以复制,这些QTL可能可以作为鉴定遗传标记和候选基因在未来被整合到遗传育种计划中以改善微生物性状。
我们还注意到,目标基因总共包含200个候选基因,这些候选基因以前在小鼠或人类研究中被报道为是与微生物性状相关的(补充表6)。其中,值得强调的有以下几点:(1)SLIT3,在英国Twins数据库中报道为与未分类的梭状芽孢杆菌科相关,与参与植物来源类固醇降解的MetaCyc途径相关,在无菌小鼠的常规化过程中,其表达在结肠隐窝中上调;(2)SLC39A8,一种具有与克罗恩病和人类肠道微生物组组成相关的多效性错义突变型的基因;和(3)NOS1,已经显示出与小鼠的体脂和肠道微生物群组成的多效性关联。我们还鉴定了已被证明与小鼠β-多样性(CSMD1、ZFAT、FRMPD1、CLEC16A、IL1R2、BANK1、PRKAG2、LHFPL3、ST5、和NXN)和肠道细菌丰度(TOX3、NAPG、DLEC1、COL19A、DDM3)相关的其他基因。Allison等人在2019年的报道中发现在原代人结肠上皮细胞(NEBL、ASAP3、ABLIM1、CUEDC2、PRRC2C、DENND1A、LAMC1、MAL2、ITGB1、CAST、A2ML1、IL7R、PCDH7、NFATC2IP、SORCS2和DNM3)中差异表达的基因也在我们的研究中被发现。此外,与人类肠道生态系统(SORCS2、LRRC32和ARAP1)的功能图谱相关的基因是我们网络中预测的目标基因。有研究表明,SORCS2与植物来源的类固醇降解途径有关。同时,位于ARAP1的遗传变异体与胆汁酸代谢相关,LRRC32与“细胞-细胞信号”转导GO条件相关。应该注意的是,这些基因中的大多数(包括关键的调控基因)都可能会被传统的单性状GWAS遗漏,GWAS对分析管道在鉴定与猪肠道微生物生态系统的多样性和组成相关的新调控基因和候选基因方面的有效性保持怀疑。宿主遗传标记的鉴定与产丁酸菌的相对丰度有关,如Faecalibacterium, Dorea, Blautia, Butyrococcus和Coprococcus(表2),我们进一步研究这些关联是否可以在猪的整体健康方面扩大,并用小猪的体重代表其健康状况和生产力。丁酸盐是肠上皮的重要能量来源,具有抗炎的潜能,可影响细胞分化并增生上皮防御屏障。事实上,已有文献记载丁酸盐对猪的生长和肠道完整性的有益作用。因此,我们主要鉴定与产丁酸盐的菌相对丰度和宿主表现相关的具有多效性效应的SNP。在校正了性别(2个水平)和批次(7个水平)的系统效应后,我们在SSC2(RS 81361511)的96,04,482 bp处发现的一个显著SNP与丁酸球菌成员的相对丰度(P = 2.43E-15)和仔猪体重(P = 0.026)相关(表4),以及与Coprococcus(rs81344777, P = 1.26E−07)和 Faecalibacterium (rs81288412, P = 1.07E−17)的相对丰度相关的两个SNP (RS 81344777,P = 1.26E-07)。值得注意的是,在这三种情况下,等位基因效应是相同的:相同的等位基因影响Butyricicoccus, Coprococcus,Faecalibacterium和仔猪体重的相对丰度。与Bergamaschi等人2020年的研究一致,我们的发现表明宿主遗传变异体的存在与有益细菌和宿主表现的相对丰度共同相关。然而,还需要更大的研究,包括实验验证和宿主(额外的表型性状)和微生物(全元基因组、元转录组)水平的替代信息来源,以充分描述宿主基因组相关微生物群落在猪生产性能、动物福利和健康中的作用。表4. 具有多效性的SNP与产丁酸菌的相对丰度和仔猪体重相关。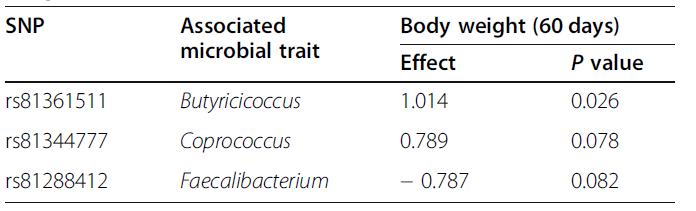
2 宿主-基因组微生物相互作用部分受宿主免疫系统调节调节因子的功能分析揭示了免疫相关途径的过度表达。据报告,IPA过度表达的途径中,多达64%(25个中的16个)的调节因子与宿主免疫反应有关(表5)。值得注意的是,该列表包括与宿主-微生物双向串扰相关的途径,如ERK/MAPK信号传导、Th1和Th2激活途径、TGF-β信号传导、Wnt/β-连环蛋白信号传导、糖皮质激素受体信号传导、VDR/ RXR激活、IL-22信号传导和芳香烃受体。
调节因子包括其他与宿主免疫系统相关的TFE3、NCOR1、SMAD4、NFE2L2、KLF7、NFATC3、TCF4、IRF2和 IKZF2等。TFE3与TFEB在调节天然免疫反应和巨噬细胞活化方面具有协同作用,而NCOR1在T细胞发育过程中对胸腺细胞的正、负选择起着至关重要的控制作用。SMAD4调节IL-2的表达,对T细胞增殖至关重要。有趣的是,根据String数据库,实验证实了SMAD4和前面提到的调节因子RUNX2、SOX9、TEF3和NCOR1之间的蛋白质相互作用。最后,调节因子还包括与生物过程相关的TFs,如炎症反应(NFE2L2,KLF7),造血(NFATC3和IKZF2),细胞介导的免疫反应(IKZF2,IRF2,NCOR1,NFATC3,RUNX2,STAT1,TCF4),体液免疫反应(IRF2,NFATC3,RUNX2,STAT1,TCF4),以及人类B细胞分化的调节(KLF14,KLF7,MTA3,STAT1)。因此,与最近在人类、小鼠和猪中的发现一致,我们证实了宿主-基因组微生物相互作用主要由宿主免疫系统决定。在本研究中,我们构建并探索了一个由3,561个基因组成的SNP-基因共关联网络,这些基因与猪肠道微生物群中31个细菌属和6个共生原生生物属的多样性和丰度有关。除了在细菌和原生生物中鉴定与α多样性相关的基因,我们的分析方法利用相关微生物性状的遗传贡献,揭示了对猪肠道微生物群分布具有多效性影响的遗传变异。研究结果还表明SNP具有多效性效应,这种多效性效应与产丁酸菌(Faecalibacterium, Butyrococcus和Coprococcus)的相对丰度和宿主表现有关。在强调调控元件的基础上,共确定了47个丰富免疫相关途径的调控因子。其中,五个调节因子在网络中突出:PRDM15、STAT1、ssc-mir-371、SOX9和RUNX2。预测靶点列表包括200个先前报道的与小鼠和人类微生物群的特征相关的候选基因,如SLIT3、SLC39A8、NOS1、IL1R2、DAB1、TOX3、SPP1、THSD7B、ELF2、PIANP、A2ML1、和IFNAR1。
综上所述,本研究认为分析管道的方法是有效的,通过利用细菌和原生生物之间的多效性和串扰在宿主-微生物组相互作用中发挥重要作用。

